6.6 EMP_enrich_analysis
Enrichment analysis is a bioinformatics approach used to determine if a set of genes or proteins is significantly enriched in specific biological pathways or functions. The database contains extensive functional information about genes and their products, as well as the roles of these genes in metabolic pathways, signaling pathways, and disease pathways. This module allows for the online reading of KEGG, GO, and Reactome database information, completing the standard steps of enrichment analysis.
6.6.1 KEGG enrichment analysis based on KO/EC annotations
The functional gene annotation results of KO/EC can be obtained during the analysis of metagenomic microbial data. Module EMP_diff_analysis can be used to retrieve differential genes, and module EMP_enrich_analysis can be utilized for KEGG enrichment analysis.
① The parameter
KEGG_Type can specify enrichment by pathway (KEGG_Type = 'KEGG') or by module (KEGG_Type = 'MKEGG').② The parameter
species defaults to using all species data as background genes (species = 'all'). Users can also specify data from a specific species as background genes for enrichment.③ The parameter
condition filters differential genes based on p-value or corrected p-value and performs enrichment.
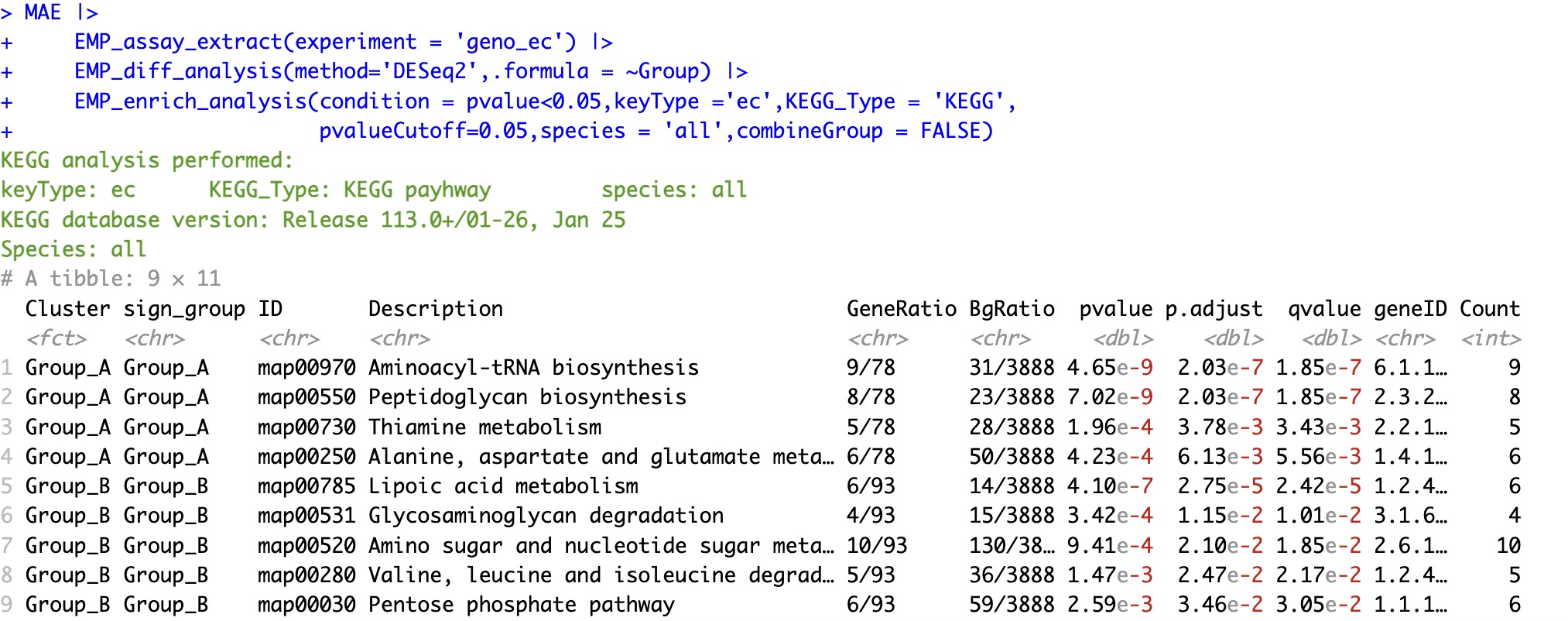
🏷️Example1:
MAE |>
EMP_assay_extract(experiment = 'geno_ec') |>
EMP_diff_analysis(method='DESeq2',.formula = ~Group) |>
EMP_enrich_analysis(condition = pvalue<0.05,keyType ='ec',KEGG_Type = 'KEGG',
pvalueCutoff=0.05,species = 'all',combineGroup = FALSE)
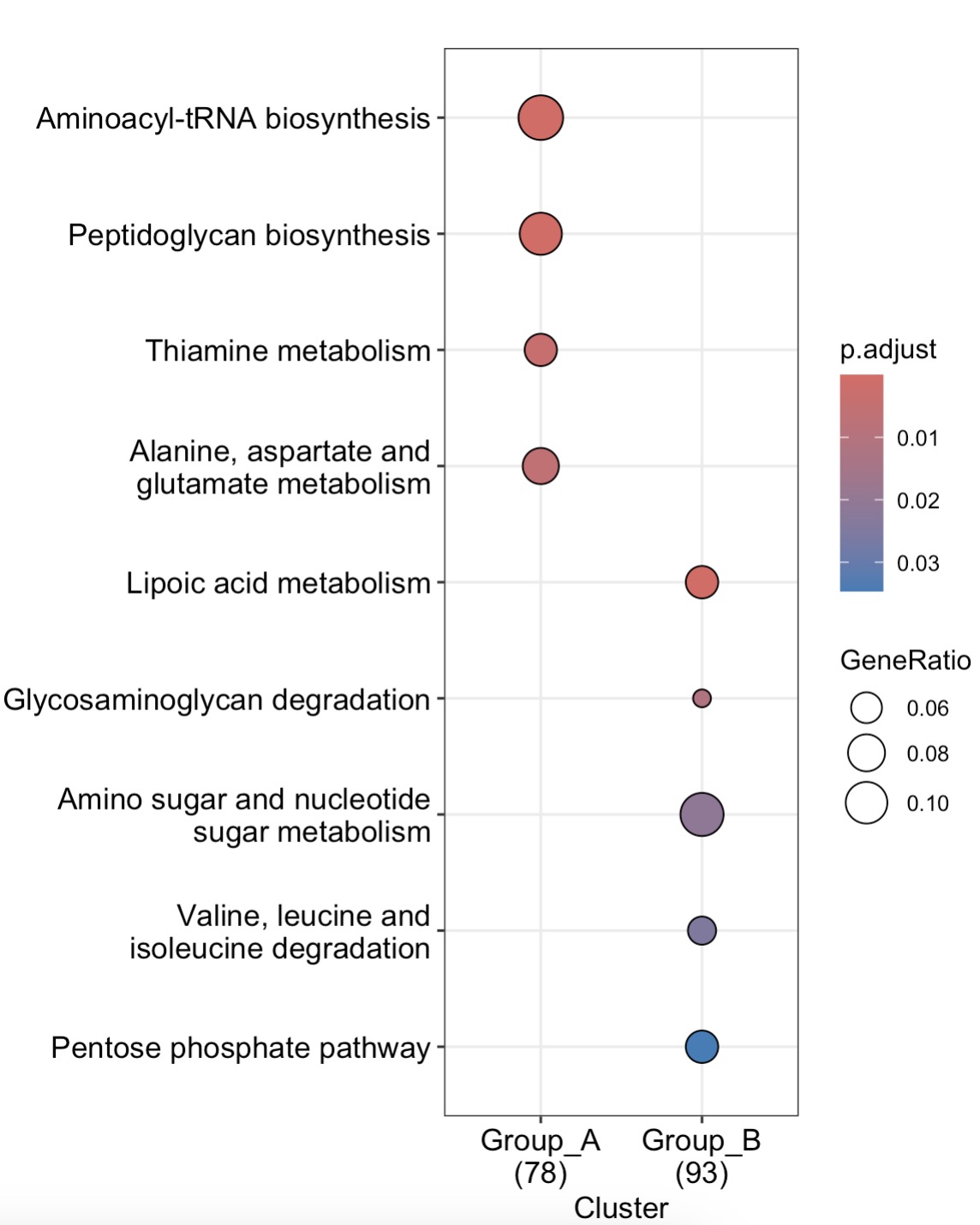
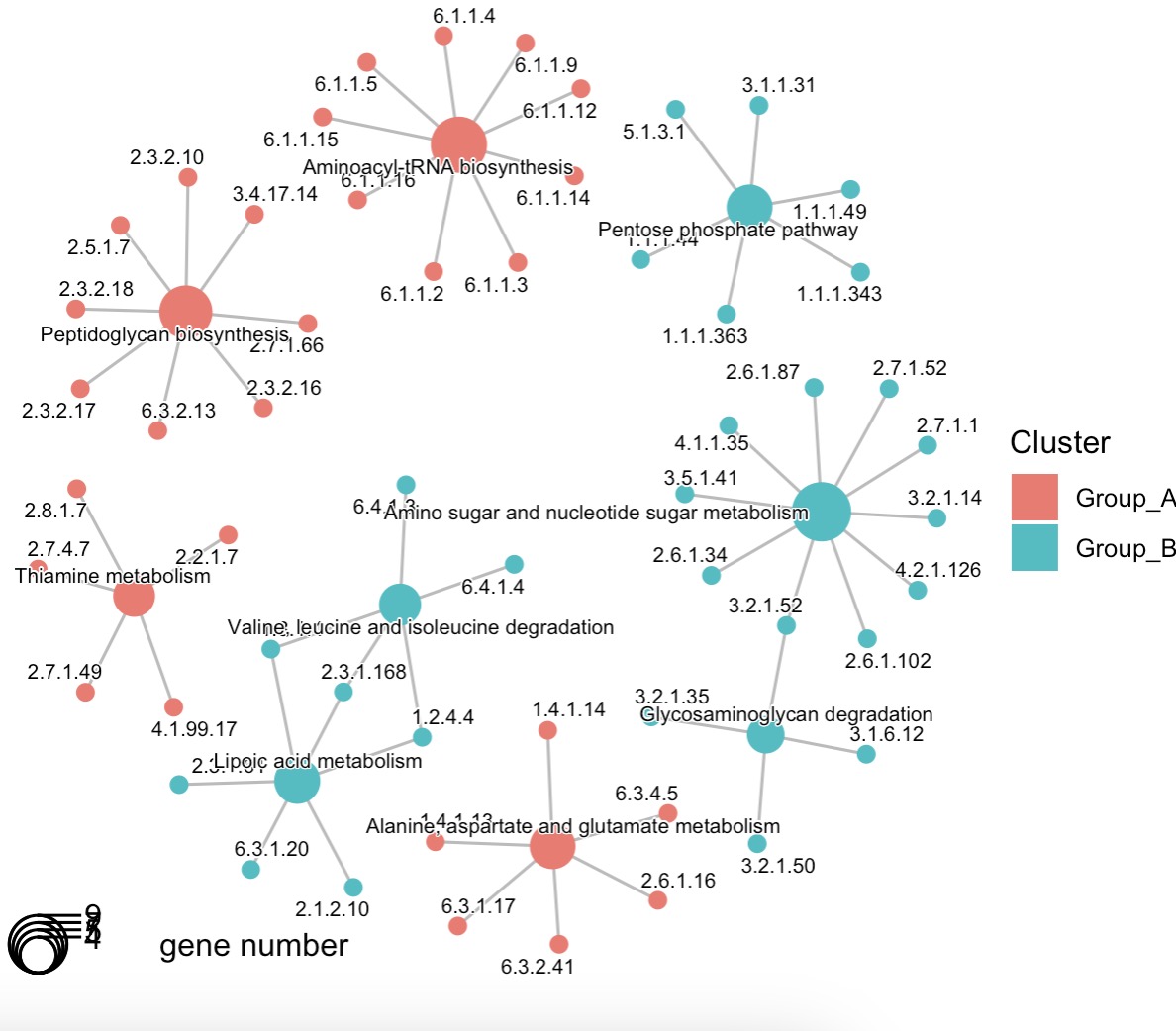
🏷️Example2:Modules EMP_enrich_dotplot and EMP_enrich_netplot inherit dotplot and cnetplot from enrichplot package, allowing for the visualization of the results from the enrichment analysis.
MAE |>
EMP_assay_extract(experiment = 'geno_ec') |>
EMP_diff_analysis(method='DESeq2',.formula = ~Group) |>
EMP_enrich_analysis(condition = pvalue<0.05,keyType ='ec',KEGG_Type = 'KEGG',
pvalueCutoff=0.05,species = 'all',combineGroup = FALSE) |>
EMP_enrich_dotplot()
MAE |>
EMP_assay_extract(experiment = 'geno_ec') |>
EMP_diff_analysis(method='DESeq2',.formula = ~Group) |>
EMP_enrich_analysis(condition = pvalue<0.05,keyType ='ec',KEGG_Type = 'KEGG',
pvalueCutoff=0.05,species = 'all',combineGroup = FALSE) |>
EMP_enrich_netplot()
6.6.2 KEGG enrichment analysis based on the gene name
This type of data is commonly found in the bulk transcriptome data of the host organism. Before analysis, it is necessary to use module EMP_feature_convert to convert gene symbols into entrezid.
🏷️Example:
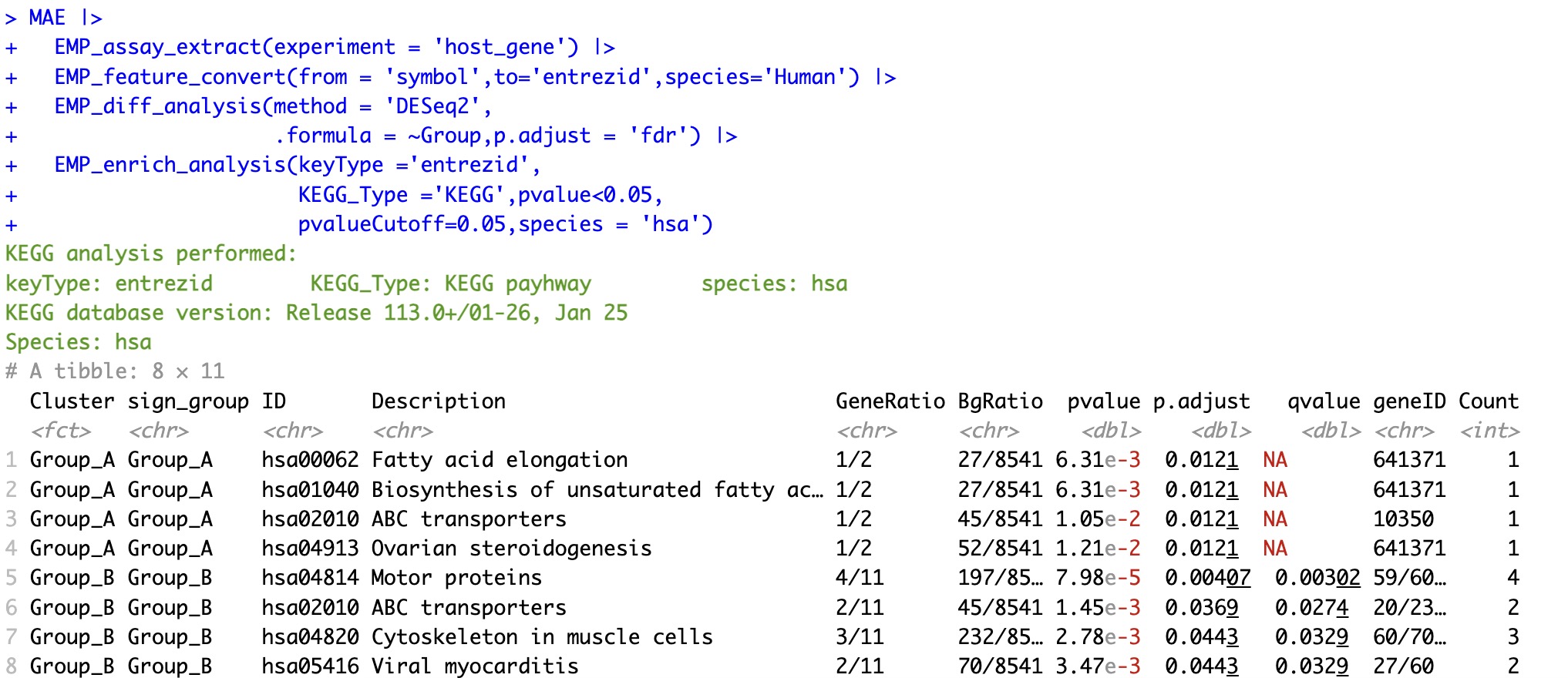
MAE |>
EMP_assay_extract(experiment = 'host_gene') |>
EMP_feature_convert(from = 'symbol',to='entrezid',species='Human') |>
EMP_diff_analysis(method = 'DESeq2',
.formula = ~Group,p.adjust = 'fdr') |>
EMP_enrich_analysis(keyType ='entrezid',
KEGG_Type ='KEGG',pvalue<0.05,
pvalueCutoff=0.05,species = 'hsa')
6.6.3 KEGG enrichment analysis based on metabolites
This type of data is mainly generated by metabolomics, and before enrichment analysis, the feature names need to be converted into compound ID.
🏷️Example:
MAE |>
EMP_assay_extract(experiment = 'untarget_metabol')|>
EMP_collapse(na_string=c('NA','null','','-'),
estimate_group = 'MS2kegg',method = 'sum',collapse_by = 'row') |>
EMP_diff_analysis(method='DESeq2',.formula = ~Group) |>
EMP_enrich_analysis(keyType ='cpd',
KEGG_Type ='KEGG',pvalue<0.05,
pvalueCutoff=0.05,species = 'all')

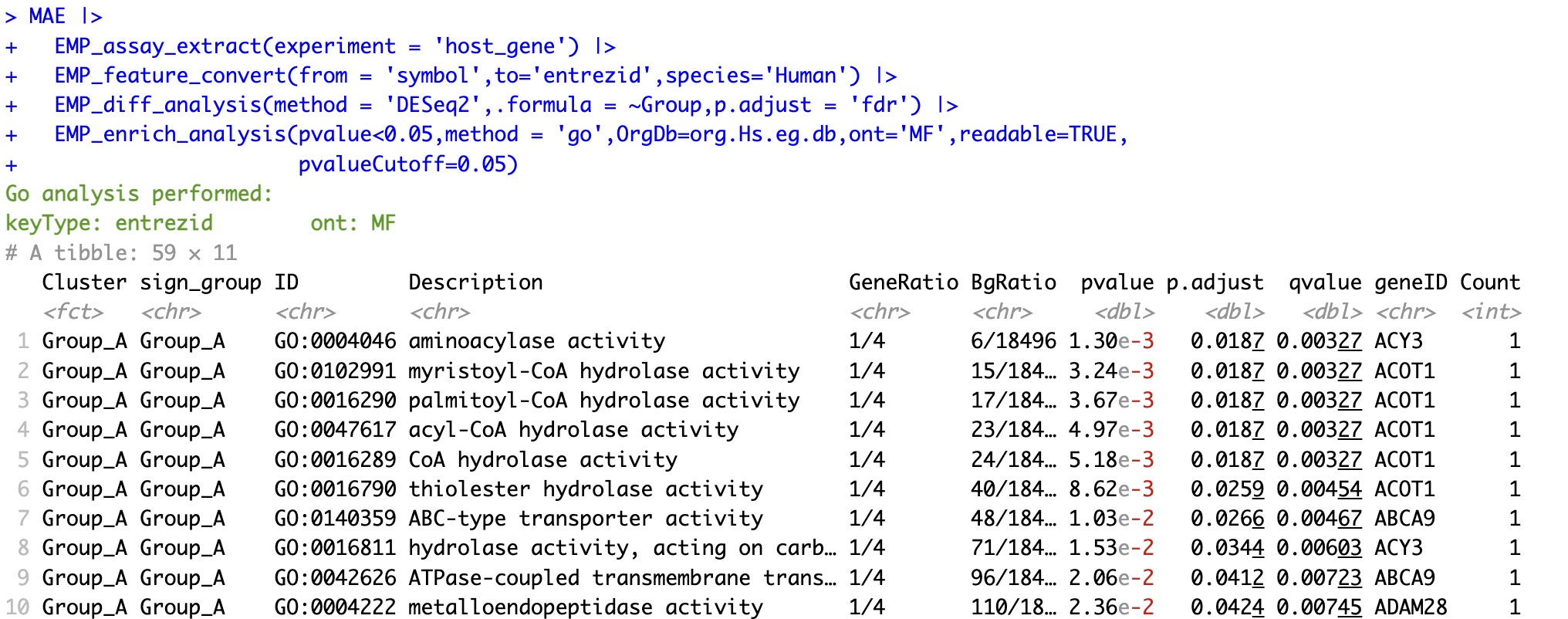
6.6.4 GO enrichment analysis based on the gene name
①The
OrgDb parameter is required in GO analysis to specify the organism.②The
ont parameter can be used to specify enrichment for BP (Biological Process), MF (Molecular Function), CC (Cellular Component), or ALL.③The
readable parameter can convert the Entrez ID in the enrichment results to symbol.
🏷️Example:
library(org.Hs.eg.db)
MAE |>
EMP_assay_extract(experiment = 'host_gene') |>
EMP_feature_convert(from = 'symbol',to='entrezid',species='Human') |>
EMP_diff_analysis(method = 'DESeq2',.formula = ~Group,p.adjust = 'fdr') |>
EMP_enrich_analysis(pvalue<0.05,method = 'go',OrgDb=org.Hs.eg.db,ont='MF',readable=TRUE,
pvalueCutoff=0.05)
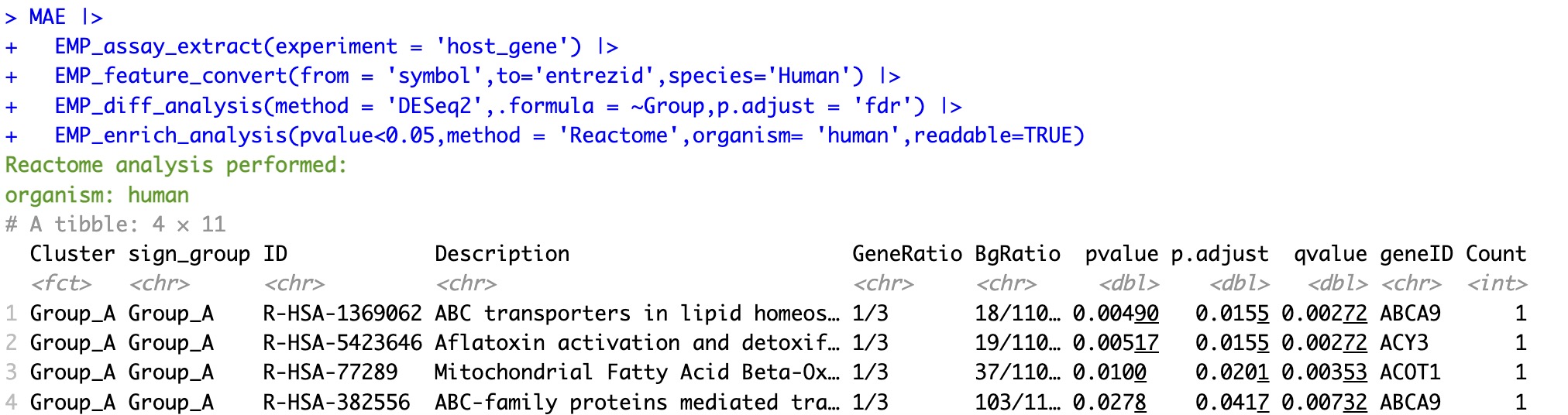
6.6.5 Reactome enrichment analysis based on the gene name
① The
organism parameter is required in Reactome analysis to specify the organism.② The readable parameter can convert the Entrez ID in the enrichment results to symbol.
🏷️Example:
MAE |>
EMP_assay_extract(experiment = 'host_gene') |>
EMP_feature_convert(from = 'symbol',to='entrezid',species='Human') |>
EMP_diff_analysis(method = 'DESeq2',.formula = ~Group,p.adjust = 'fdr') |>
EMP_enrich_analysis(pvalue<0.05,method = 'Reactome',organism= 'human',readable=TRUE)
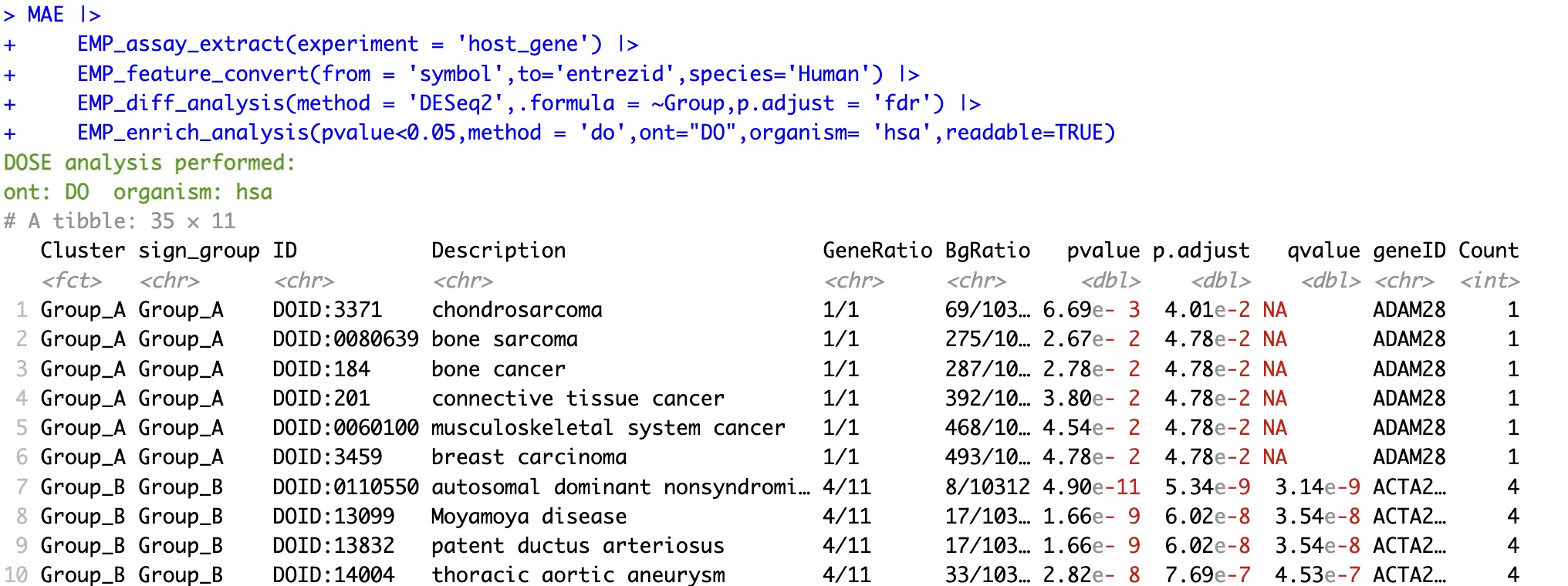
6.6.6 DOSE enrichment analysis based on the gene name
① The
organism parameter is required in DOSE analysis to specify the organism. Please note that the syntax for this parameter differs from that in Reactome.② The
ont parameter supports the HDO, HPO and MPO mode.
🏷️Example:
MAE |>
EMP_assay_extract(experiment = 'host_gene') |>
EMP_feature_convert(from = 'symbol',to='entrezid',species='Human') |>
EMP_diff_analysis(method = 'DESeq2',.formula = ~Group,p.adjust = 'fdr') |>
EMP_enrich_analysis(pvalue<0.05,method = 'do',ont="HDO",organism= 'hsa',readable=TRUE)